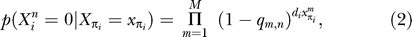I did not read this abst. I don't understand it.
Protein Molecular Function Prediction by Bayesian Phylogenomics
We present a statistical graphical model to infer specific molecular function for unannotated protein sequences using homology.
"DNA annotation or genome annotation is the process of (a) identifying the locations/segments of genes, coding regions and other specific locations that are of imporatance in a DNA sequence or genome and (b) associating relevant information with those locations/segments (e.g. determining what the identified genes do). Once a genome is sequenced, it needs to be annotated in order to make sense of it."
DNA Sequence Annotation Studio
An annotated sequence is a sequence for which the function encoded is known. Unannotated, unknown.
Based on phylogenomic principles, SIFTER (Statistical Inference of Function Through Evolutionary Relationships) accurately predicts molecular function for members of a protein family given a reconciled phylogeny and available function annotations, even when the data are sparse or noisy.
A protein family is usually a set of proteins with a similar sequence, structure and function. The annotated sequences of some of the other members within the protein family is fed into the program and it is asked to find the function of the sequences (annotate).
Our method produced specific and consistent molecular function predictions across 100 Pfam families in comparison to the Gene Ontology annotation database, BLAST, GOtcha, and Orthostrapper.
BLAST, GOtcha, and Orthostrapper are databases which already contain an annotation of the sequences being fed in. They can be used as references to check the accuracy of SIFTER and the new method.
The new method is a mathematical function, a sort of algorithm.
"Let Xi denote the Boolean vector of candidate molecular function annotations for node i and let denote the mth component of this vector. Let M denote the number of components of this vector. Let πi denote the immediate ancestor of node i in the phylogeny, so that denotes the annotation vector at the ancestor. We define the transition probability associated with the branch from πi to i as follows"
Note
Protein Molecular Function Prediction by Bayesian Phylogenomics
We chose a statistical model known as a loglinear model for the model of function evolution. We make no claims for any theoretical justification of this model. It is simply a phenomenological model that captures in broad outlines some of the desiderata of an evolutionary model for function and has worked well in practice in our phylogenomic setting.
We performed a more detailed exploration of functional predictions on the adenosine-5′-monophosphate/adenosine deaminase family and the lactate/malate dehydrogenase family, in the former case comparing the predictions against a gold standard set of published functional characterizations.
Protein families.
The performance of SIFTER is analysed through referencing published functional characterizations (or annotated sequences).
Given function annotations for 3% of the proteins in the deaminase family, SIFTER achieves 96% accuracy in predicting molecular function for experimentally characterized proteins as reported in the literature.
SIFTER is given the annotated sequences of 3% of the proteins within the family and is asked to find the function of the other members of the family. It scores 96%.
The accuracy of SIFTER on this dataset is a significant improvement over other currently available methods such as BLAST (75%), GeneQuiz (64%), GOtcha (89%), and Orthostrapper (11%).
Better than others.
We also experimentally characterized the adenosine deaminase from Plasmodium falciparum, confirming SIFTER's prediction.
Plasmodium falciparum contains members of the family and SIFTER annotated the sequences through the method just outlined.
The results illustrate the predictive power of exploiting a statistical model of function evolution in phylogenomic problems.
Biologists are joyful. It shows that when some members of the family are annotated and fed in, the function of the other members, which often times have a similar structure, sequence and function, can be statistically deduced.
A software implementation of SIFTER is available from the authors.
Marketing product.
But, what if I say a creationist can use "the same" method? Does that make this creationist not qualified to be a creationist?
It's funny you should ask that. We use the Orchard.
Testing the Orchard Model and the NCSE's Claims of "Nested Patterns" Supporting a "Tree of Life" - Evolution News & Views
There was also the case where an evolutionary relationship was surmised but it failed in real time and showed constraints.
Michael Behe's Blog - Uncommon Descent - Part 2
In the paper Bridgham et al (2009) continue their earlier work on steroid hormone receptor evolution. Previously they had constructed in the laboratory a protein which they inferred to be the ancestral sequence of two modern hormone receptors abbreviated GR and MR (Bridgham et al 2006). They then showed that if they changed two amino acid residues in the inferred ancestral receptor protein into ones which occur in GR, they could change its binding specificity somewhat in the direction of modern GR’s specificity. (All the work was done on molecules in the laboratory. No measurements were made of the selective value of the changes in real organisms in nature. Thus any relevance to actual biology is speculative.) They surmised that a gene duplication plus sequence diversification could have given rise to MR and GR. As I wrote in a comment at the time (
CSC - Michael Behe On The Theory of Irreducible Complexity ), that was interesting work, and the conclusion was reasonable, but the result was exceedingly modest and well within the boundaries that an intelligent design proponent like myself would ascribe to Darwinian processes. After all, the starting point was a protein which binds several steroid hormones, and the ending point was a slightly different protein that binds the same steroid hormones with slightly different strengths. How hard could that be?
Well, it turns out that Darwinian evolution can have a lot of trouble accomplishing even that simple task, or at least its opposite. In the new paper the authors try the reverse experiment. They begin with the more modern hormone receptor (which is more restrictive in the steroids it binds) and ask whether a Darwinian process could get the ancestral activity back (which is more permissive). Their answer is no, it couldn’t. They show that a handful of amino acid residues in the more recent receptor would first have to be changed before it could act as the ancestral form is supposed to have done, and that is very unlikely to occur. In other words, the new starting point is also a protein which binds a steroid hormone, and the new desired ending point is also a slightly different protein that binds steroid hormones. How hard could that be? But it turns out that Darwinian processes can’t reach it, because several amino acids would have to be altered before the target activity kicked in.