Do humans and apes share a common ancestor? What evidence supports this hypothesis? Even more importantly, what type of observations would be consistent with common ancestry, and which observations would be inconsistent with common ancestry?
Let’s move away from biology for just a moment. Let’s look at languages, specifically the Romance Languages (e.g., French, Spanish, Italian). These languages share a common ancestor in Vulgar Latin. When the Roman Empire fell their old territories, in which Latin was spoken, started to take on their own word usages, dialects, and finally own languages. The “evolution” of the Romance Languages looks something like this:

(source: http://en.wikipedia.org/wiki/Romance_languages ).
So how do we know that these languages are related, or rather share common ancestry? Anyone who has studied French, Spanish, or Italian will notice that some words are pronounced or spelled very similarly in each of the languages. It would seem very unlikely that two isolated cultures would independently arrive at a very similar word for the same object or action, wouldn’t it? Are not commonalities better explained by a common source than unrelated sources?
Common ancestry, through morphology and genetics, works in the same way. When we see two features that are nearly identical shared between two species it is hard to imagine that these arose independently. It is much more likely that these arose from an ancestor shared by the two species. Not only that, but the pattern of similarities should match the same tree structure that the Romance Languages fall into. This pattern is called a nested hierarchy, and it is the most powerful evidence of common ancestry. One genetic feature of primates that falls into a nested hierarchy are endogenous retroviruses (ERV’s). This will be the focus of this post.
ERV’s are retroviral genomes that have become part of the host genome. When a retrovirus attaches and injects it’s RNA into the host cell that RNA is reverse transcribed into DNA. That DNA is inserted into the host genome which then hijacks the host’s cellular processes to make more viral particles. Most of the time this occurs in somatic cells, the cells that carry out processes that keep us alive. However, once in a while a retrovirus will insert into a germ line cell which are the cells used in reproduction (ie sperm and eggs). If this insertion is somehow imperfect the virus can not replicate. The offspring produced from these germ line cells will carry a copy of retrovirus in every cell in their body. It effectively becomes part of their genome which they pass on to their offspring.
So how do we detect ERV’s in genomes? Retroviruses have very specific genes which include a reverse polymerase (pol), group specific antigen (gag), and an envelope gene (env). These genes are flanked by long terminal repeats (LTR’s) which are responsible for turning on transcription of the viral genes. Recently inserted ERV’s will have most or all of these genes. However, over time there is a high probability that the repeat regions in the flanking LTR’s will recombine with each other leaving a solo LTR in the host genome (note: repeat regions are prone to recombination).
Retroviruses insert into the host genome among many, many insertion sites. Retroviruses also have different preferences of insertion sites. For example, the table below outlines where HIV, ASLV, and MLV insert into identical human genomes. As you can see, there are thousands of insertion sites spread over the entire genome.

Caption: The human chromosomes are shown numbered. HIV integration sites from all datasets in Table 1 are shown as blue “lollipops”; MLV integration sites are shown in lavender; and ASLV integration sites are shown in green. Transcriptional activity is shown by the red shading on each of the chromosomes (derived from quantification of nonnormalized EST libraries, see text). Centromeres, which are mostly unsequenced, are shown as grey rectangles. (source: http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=15314653 ).
If you read the paper you will learn that these viruses prefer areas such as CpG islands and regions upstream of heavily transcribed DNA. These regions are large enough to supply thousands of possible insertion sites for each type of virus.
Much like commonalities between Romance Languages, it is exceedingly unlikely that an ERV shared at the same position in the genome between two species is due to two independent events. Therefore, if chimps and humans share an ERV at the same genomic position it is most likely that the retrovirus inserted into the genome of a common ancestor. If there are thousands of ERV’s shared in this way, it is almost certainly likely that they share a common ancestor. For this same reason, you, your parents, and your siblings share thousands of ERV’s at the same genomic position without ever being infected by a virus.
Luckily for us, both the human and chimp genomes have been sequenced. The results? Out of the ~200,000 ERV’s present in the human genome only 82 are not found in the chimp genome. In the chimp genome, only 280 can not be found in the human genome. That means that hundreds of thousands of ERV’s are shared between humans (sources: the human genome paper and the chimp genome paper referenced at the end of the post). The 360 total ERV’s that are not shared represent insertions since the chimp and human lineages diverged. This is also evidenced by the fact that these unshared ERV’s often contain env, gag, and pol genes which is consistent with a recent insertion event.
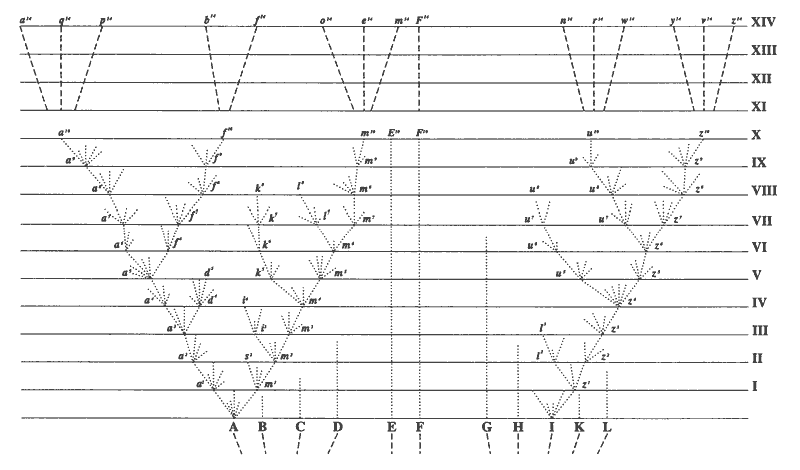
But what about the nested hierarchy that I mentioned above? If we pull back the zoom lens and look at all primates we see exactly that pattern of shared ERV’s.
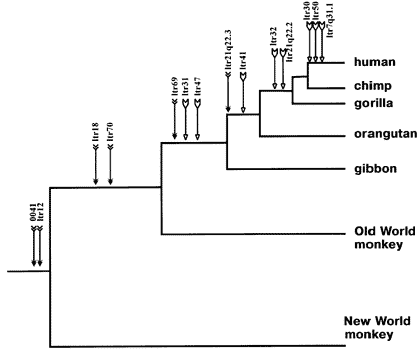
Every species to the right of each arrow along the tree has those ERV’s at the same genomic position, the exact pattern that common ancestry would produce.
According to evolution there should also be another nested hierarchy created by the sequence of the shared ERV’s themselves. When two lineages diverge they will accrue mutations that are specific to that lineage. ERV’s accrue mutations just like every other piece of DNA in the genome. Using our Romance Languages analogy, each generation adds their own changes in language so that over time the languages diverge but still keep their commonalities that demonstrate their shared ancestry. The amount of time since the divergence directly correlates with the amount of difference between the languages. Therefore, if two species share a distant common ancestor there should be more sequence difference between the same shared ERV. A more recent ancestor will result in a sequence that is more alike. Therefore, a comparison of shared ERV sequences should yield the same nested hierarchy, and it does (source: http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=10468595 ).
In summary, ERV’s are very strong evidence for common ancestry between chimps and humans, and for all primates for that matter. If they were not related by common ancestry we would not expect to see a nested hierarchy, the pattern that evolution and common ancestry are expected to produce. Anyone wishing to refute this argument needs to explain this fundamental observation, the presence of a nested hierarchy among shared characteristics. Not only does the genomic placement of the ERV’s produce a nested hierarchy, so does the sequence of the shared ERV’s and the same nested hierarchy at that. It is evidence like this which leads scientists to accept the hypothesis that chimps and humans share a common ancestor.
Human genome paper: http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&list_uids=11237011&dopt=Abstract
Chimp genome paper: http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&list_uids=16136131&dopt=Abstract
Let’s move away from biology for just a moment. Let’s look at languages, specifically the Romance Languages (e.g., French, Spanish, Italian). These languages share a common ancestor in Vulgar Latin. When the Roman Empire fell their old territories, in which Latin was spoken, started to take on their own word usages, dialects, and finally own languages. The “evolution” of the Romance Languages looks something like this:
(source: http://en.wikipedia.org/wiki/Romance_languages ).
So how do we know that these languages are related, or rather share common ancestry? Anyone who has studied French, Spanish, or Italian will notice that some words are pronounced or spelled very similarly in each of the languages. It would seem very unlikely that two isolated cultures would independently arrive at a very similar word for the same object or action, wouldn’t it? Are not commonalities better explained by a common source than unrelated sources?
Common ancestry, through morphology and genetics, works in the same way. When we see two features that are nearly identical shared between two species it is hard to imagine that these arose independently. It is much more likely that these arose from an ancestor shared by the two species. Not only that, but the pattern of similarities should match the same tree structure that the Romance Languages fall into. This pattern is called a nested hierarchy, and it is the most powerful evidence of common ancestry. One genetic feature of primates that falls into a nested hierarchy are endogenous retroviruses (ERV’s). This will be the focus of this post.
ERV’s are retroviral genomes that have become part of the host genome. When a retrovirus attaches and injects it’s RNA into the host cell that RNA is reverse transcribed into DNA. That DNA is inserted into the host genome which then hijacks the host’s cellular processes to make more viral particles. Most of the time this occurs in somatic cells, the cells that carry out processes that keep us alive. However, once in a while a retrovirus will insert into a germ line cell which are the cells used in reproduction (ie sperm and eggs). If this insertion is somehow imperfect the virus can not replicate. The offspring produced from these germ line cells will carry a copy of retrovirus in every cell in their body. It effectively becomes part of their genome which they pass on to their offspring.
So how do we detect ERV’s in genomes? Retroviruses have very specific genes which include a reverse polymerase (pol), group specific antigen (gag), and an envelope gene (env). These genes are flanked by long terminal repeats (LTR’s) which are responsible for turning on transcription of the viral genes. Recently inserted ERV’s will have most or all of these genes. However, over time there is a high probability that the repeat regions in the flanking LTR’s will recombine with each other leaving a solo LTR in the host genome (note: repeat regions are prone to recombination).
Retroviruses insert into the host genome among many, many insertion sites. Retroviruses also have different preferences of insertion sites. For example, the table below outlines where HIV, ASLV, and MLV insert into identical human genomes. As you can see, there are thousands of insertion sites spread over the entire genome.
Caption: The human chromosomes are shown numbered. HIV integration sites from all datasets in Table 1 are shown as blue “lollipops”; MLV integration sites are shown in lavender; and ASLV integration sites are shown in green. Transcriptional activity is shown by the red shading on each of the chromosomes (derived from quantification of nonnormalized EST libraries, see text). Centromeres, which are mostly unsequenced, are shown as grey rectangles. (source: http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=15314653 ).
If you read the paper you will learn that these viruses prefer areas such as CpG islands and regions upstream of heavily transcribed DNA. These regions are large enough to supply thousands of possible insertion sites for each type of virus.
Much like commonalities between Romance Languages, it is exceedingly unlikely that an ERV shared at the same position in the genome between two species is due to two independent events. Therefore, if chimps and humans share an ERV at the same genomic position it is most likely that the retrovirus inserted into the genome of a common ancestor. If there are thousands of ERV’s shared in this way, it is almost certainly likely that they share a common ancestor. For this same reason, you, your parents, and your siblings share thousands of ERV’s at the same genomic position without ever being infected by a virus.
Luckily for us, both the human and chimp genomes have been sequenced. The results? Out of the ~200,000 ERV’s present in the human genome only 82 are not found in the chimp genome. In the chimp genome, only 280 can not be found in the human genome. That means that hundreds of thousands of ERV’s are shared between humans (sources: the human genome paper and the chimp genome paper referenced at the end of the post). The 360 total ERV’s that are not shared represent insertions since the chimp and human lineages diverged. This is also evidenced by the fact that these unshared ERV’s often contain env, gag, and pol genes which is consistent with a recent insertion event.
But what about the nested hierarchy that I mentioned above? If we pull back the zoom lens and look at all primates we see exactly that pattern of shared ERV’s.
Every species to the right of each arrow along the tree has those ERV’s at the same genomic position, the exact pattern that common ancestry would produce.
According to evolution there should also be another nested hierarchy created by the sequence of the shared ERV’s themselves. When two lineages diverge they will accrue mutations that are specific to that lineage. ERV’s accrue mutations just like every other piece of DNA in the genome. Using our Romance Languages analogy, each generation adds their own changes in language so that over time the languages diverge but still keep their commonalities that demonstrate their shared ancestry. The amount of time since the divergence directly correlates with the amount of difference between the languages. Therefore, if two species share a distant common ancestor there should be more sequence difference between the same shared ERV. A more recent ancestor will result in a sequence that is more alike. Therefore, a comparison of shared ERV sequences should yield the same nested hierarchy, and it does (source: http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=10468595 ).
In summary, ERV’s are very strong evidence for common ancestry between chimps and humans, and for all primates for that matter. If they were not related by common ancestry we would not expect to see a nested hierarchy, the pattern that evolution and common ancestry are expected to produce. Anyone wishing to refute this argument needs to explain this fundamental observation, the presence of a nested hierarchy among shared characteristics. Not only does the genomic placement of the ERV’s produce a nested hierarchy, so does the sequence of the shared ERV’s and the same nested hierarchy at that. It is evidence like this which leads scientists to accept the hypothesis that chimps and humans share a common ancestor.
Human genome paper: http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&list_uids=11237011&dopt=Abstract
Chimp genome paper: http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&list_uids=16136131&dopt=Abstract