I originally wrote this post in the formal debates section, but it appears that my opponent is not going to be back to debate it. Since I spent time on the post, I thought I would post it here for others to comment.
I will be citing genetic evidence that humans share a common ancestor with chimps and other primates, much of which I have described elsewhere here at CF. However, I have yet to find anyone who can directly debate these points.
The genetic evidence I am talking about is endogenous retroviruses (ERVs). They are called retroviruses because they have a genome made of RNA, and they use reverse transcriptase to copy that RNA into DNA (i.e backwards, or retro). This DNA viral genome is then inserted into the host genome, hence the usage of the term “endogenous”. If this viral insertion happens in an egg or sperm, the offspring that comes from those gametes will have a permanent copy of that viral genome in its DNA which it can also pass on to its offspring. As it turns out, the human genome contains 208,000 ERVs, making up about 4.5% of the total human genome (ERV-classI-III in the table below, excluding MaLR from list).
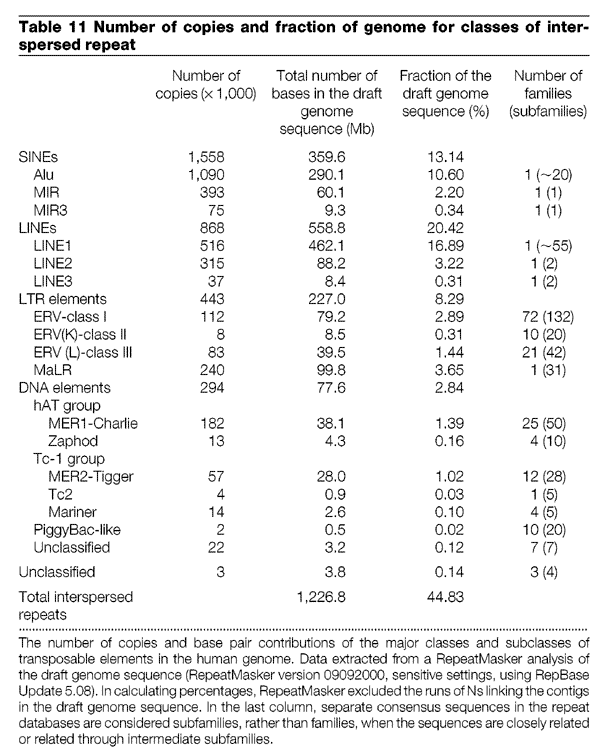
Human Genome paper 2001: http://www.nature.com/nature/journal/v409/n6822/full/409860a0.html
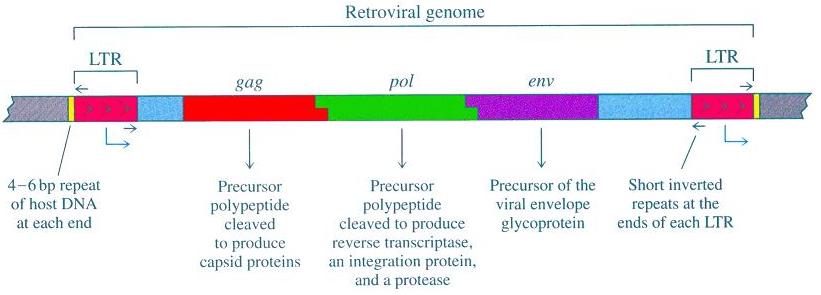
We identify them as retroviruses because ERVs have the usual complement of viral genes flanked by long tandem repeats (LTRs) at the beginning and end of the ERV.
On an interesting note, scientists have aligned these different ERVs, found the consensus sequence, and reconstructed a model ancestral viral genome. What they got was a functional retrovirus:
“Here, we derived in silico the sequence of the putative ancestral “progenitor” element of one of the most recently amplified family—the HERV-K family—and constructed it. This element, Phoenix, produces viral particles that disclose all of the structural and functional properties of a bona-fide retrovirus, can infect mammalian, including human, cells, and integrate with the exact signature of the presently found endogenous HERV-K progeny.”
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1665638/
One important thing to note is that they had to remove the mutations from these sequences in order to get a functional retrovirus. This is NOT consistent with a scenario where widely shared ERVs are the source for new retroviruses. If ERVs were producing new retroviruses then you wouldn’t need to remove the mutations as part of a consensus sequence in order to get a functional retrovirus. The evidence is clearly in favor of ERVs being the product of retroviral insertions in the past which have accumulated mutations since insertion.
So why can ERVs be used as genetic markers, and a test for common ancestry? As stated earlier, part of the viral life cycle is insertion into the host genome. The human haploid genome is around 3 billion bases, as is the genome of other ape species. That’s 3 billion possible places where these retroviruses can insert. When viruses insert into the genome, they don’t insert at just one base. They insert all over the place. In this study, scientists infected cells with three different retroviruses: MLV, HIV, and ASLV. After infection, they mapped where the viruses inserted into the host genome. Below is map of where those viruses inserted, broken down in the 23 human autosomal chromosomes and the X chromosome.

Relationship between Integration Sites and Transcriptional Intensity in the Human Genome
The human chromosomes are shown numbered. HIV integration sites from all datasets in Table 1 are shown as blue “lollipops”; MLV integration sites are shown in lavender; and ASLV integration sites are shown in green. Transcriptional activity is shown by the red shading on each of the chromosomes (derived from quantification of nonnormalized EST libraries, see text). Centromeres, which are mostly unsequenced, are shown as grey rectangles.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC509299/
As everyone can see, the viruses inserted all over the place, into all chromosomes.
So why is this important? As one paper put it:
“Given the size of vertebrate genomes (>1 × 10^9 bp) and the random nature of retroviral integration (22, 23), multiple integrations (and subsequent fixation) of ERV loci at precisely the same location are highly unlikely (24). Therefore, an ERV locus shared by two or more species is descended from a single integration event and is proof that the species share a common ancestor into whose germ line the original integration took place (14).”
http://www.pnas.org/content/96/18/10254.full
The chances of two viral insertions occurring at the same base in two species is extremely low, especially when we are talking about 208,000 ERVs.
What do we see when we compare the human and chimp genomes. Are these ERVs found at the same base in both species, or at different bases? Here are the results from the chimp genome paper where they compared the chimp and human ERVs.

http://www.nature.com/nature/journal/v437/n7055/full/nature04072.html
In ERV class 1 and 2, only a total of 82 human ERVs were not found at the same place in the chimp genome (i.e. lineage specific insertions). This means that more than 99.9% of the human ERVs are found at the same base in the chimp genome, which is nearly all 208,000 insertions. This can’t be explained by separate infections in the human and chimp lineage. This clearly points to a single insertion occurring in a common ancestor, and that ERV being passed down in both lineages.
One of the common rebuttals to this evidence is that there are insertional hotspots. One of the oft cited papers is this one, where they report a 280 fold increase in the insertion rate for a specific sequence of DNA:
https://www.researchgate.net/public..._for_avian_retrovirus_DNA_integration_in_vivo
What the creationists don’t tell you is the base probability that is seeing a 280 fold increase. If I bought 280 Powerball tickets instead of 1 I would increase my odds of winning by 280. Does this mean that I will win 99.9% of the time? Absolutely not. The base probability of winning the Powerball lottery is about 1 in 175 million. Increasing my odds of winning by 280 will not come close to guaranteeing a win. So what is the base probability for these hotpsots?

The base probability is 2-3 in 10 million insertions. A 280 fold increase would be about 900 insertions in the hotspot for every 10 million total insertions. If hotspots were responsible for finding ERVs at the same base in each genome, then much less than 1% of ERVs should be found at the same location. Instead, more than 99.9% of ERVs are found at the same spot in the human and chimp genome. Hotspots can’t be the cause. That leaves us with common ancestry.
There is also an additional layer of genetic evidence involving ERVs which can be discussed in further posts if necessary. For those who want to read ahead . . .
“Third, sequence divergence between the LTRs at the ends of a given provirus provides an important and unique source of phylogenetic information. The LTRs are created during reverse transcription to regenerate cis-acting elements required for integration and transcription. Because of the mechanism of reverse transcription, the two LTRs must be identical at the time of integration, even if they differed in the precursor provirus (Fig. 1A). Over time, they will diverge in sequence because of substitutions, insertions, and deletions acquired during cellular DNA replication. Although it has been noted that the divergence between the two LTRs of an ERV can serve as a molecular clock (8, 15, 18, 25), there are no reported prior attempts to utilize the LTRs of individual ERV loci as a source of phylogenetic signal.”
http://www.pnas.org/content/96/18/10254.full
I will be citing genetic evidence that humans share a common ancestor with chimps and other primates, much of which I have described elsewhere here at CF. However, I have yet to find anyone who can directly debate these points.
The genetic evidence I am talking about is endogenous retroviruses (ERVs). They are called retroviruses because they have a genome made of RNA, and they use reverse transcriptase to copy that RNA into DNA (i.e backwards, or retro). This DNA viral genome is then inserted into the host genome, hence the usage of the term “endogenous”. If this viral insertion happens in an egg or sperm, the offspring that comes from those gametes will have a permanent copy of that viral genome in its DNA which it can also pass on to its offspring. As it turns out, the human genome contains 208,000 ERVs, making up about 4.5% of the total human genome (ERV-classI-III in the table below, excluding MaLR from list).
Human Genome paper 2001: http://www.nature.com/nature/journal/v409/n6822/full/409860a0.html
We identify them as retroviruses because ERVs have the usual complement of viral genes flanked by long tandem repeats (LTRs) at the beginning and end of the ERV.
On an interesting note, scientists have aligned these different ERVs, found the consensus sequence, and reconstructed a model ancestral viral genome. What they got was a functional retrovirus:
“Here, we derived in silico the sequence of the putative ancestral “progenitor” element of one of the most recently amplified family—the HERV-K family—and constructed it. This element, Phoenix, produces viral particles that disclose all of the structural and functional properties of a bona-fide retrovirus, can infect mammalian, including human, cells, and integrate with the exact signature of the presently found endogenous HERV-K progeny.”
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1665638/
One important thing to note is that they had to remove the mutations from these sequences in order to get a functional retrovirus. This is NOT consistent with a scenario where widely shared ERVs are the source for new retroviruses. If ERVs were producing new retroviruses then you wouldn’t need to remove the mutations as part of a consensus sequence in order to get a functional retrovirus. The evidence is clearly in favor of ERVs being the product of retroviral insertions in the past which have accumulated mutations since insertion.
So why can ERVs be used as genetic markers, and a test for common ancestry? As stated earlier, part of the viral life cycle is insertion into the host genome. The human haploid genome is around 3 billion bases, as is the genome of other ape species. That’s 3 billion possible places where these retroviruses can insert. When viruses insert into the genome, they don’t insert at just one base. They insert all over the place. In this study, scientists infected cells with three different retroviruses: MLV, HIV, and ASLV. After infection, they mapped where the viruses inserted into the host genome. Below is map of where those viruses inserted, broken down in the 23 human autosomal chromosomes and the X chromosome.
Relationship between Integration Sites and Transcriptional Intensity in the Human Genome
The human chromosomes are shown numbered. HIV integration sites from all datasets in Table 1 are shown as blue “lollipops”; MLV integration sites are shown in lavender; and ASLV integration sites are shown in green. Transcriptional activity is shown by the red shading on each of the chromosomes (derived from quantification of nonnormalized EST libraries, see text). Centromeres, which are mostly unsequenced, are shown as grey rectangles.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC509299/
As everyone can see, the viruses inserted all over the place, into all chromosomes.
So why is this important? As one paper put it:
“Given the size of vertebrate genomes (>1 × 10^9 bp) and the random nature of retroviral integration (22, 23), multiple integrations (and subsequent fixation) of ERV loci at precisely the same location are highly unlikely (24). Therefore, an ERV locus shared by two or more species is descended from a single integration event and is proof that the species share a common ancestor into whose germ line the original integration took place (14).”
http://www.pnas.org/content/96/18/10254.full
The chances of two viral insertions occurring at the same base in two species is extremely low, especially when we are talking about 208,000 ERVs.
What do we see when we compare the human and chimp genomes. Are these ERVs found at the same base in both species, or at different bases? Here are the results from the chimp genome paper where they compared the chimp and human ERVs.
http://www.nature.com/nature/journal/v437/n7055/full/nature04072.html
In ERV class 1 and 2, only a total of 82 human ERVs were not found at the same place in the chimp genome (i.e. lineage specific insertions). This means that more than 99.9% of the human ERVs are found at the same base in the chimp genome, which is nearly all 208,000 insertions. This can’t be explained by separate infections in the human and chimp lineage. This clearly points to a single insertion occurring in a common ancestor, and that ERV being passed down in both lineages.
One of the common rebuttals to this evidence is that there are insertional hotspots. One of the oft cited papers is this one, where they report a 280 fold increase in the insertion rate for a specific sequence of DNA:
https://www.researchgate.net/public..._for_avian_retrovirus_DNA_integration_in_vivo
What the creationists don’t tell you is the base probability that is seeing a 280 fold increase. If I bought 280 Powerball tickets instead of 1 I would increase my odds of winning by 280. Does this mean that I will win 99.9% of the time? Absolutely not. The base probability of winning the Powerball lottery is about 1 in 175 million. Increasing my odds of winning by 280 will not come close to guaranteeing a win. So what is the base probability for these hotpsots?
The base probability is 2-3 in 10 million insertions. A 280 fold increase would be about 900 insertions in the hotspot for every 10 million total insertions. If hotspots were responsible for finding ERVs at the same base in each genome, then much less than 1% of ERVs should be found at the same location. Instead, more than 99.9% of ERVs are found at the same spot in the human and chimp genome. Hotspots can’t be the cause. That leaves us with common ancestry.
There is also an additional layer of genetic evidence involving ERVs which can be discussed in further posts if necessary. For those who want to read ahead . . .
“Third, sequence divergence between the LTRs at the ends of a given provirus provides an important and unique source of phylogenetic information. The LTRs are created during reverse transcription to regenerate cis-acting elements required for integration and transcription. Because of the mechanism of reverse transcription, the two LTRs must be identical at the time of integration, even if they differed in the precursor provirus (Fig. 1A). Over time, they will diverge in sequence because of substitutions, insertions, and deletions acquired during cellular DNA replication. Although it has been noted that the divergence between the two LTRs of an ERV can serve as a molecular clock (8, 15, 18, 25), there are no reported prior attempts to utilize the LTRs of individual ERV loci as a source of phylogenetic signal.”
http://www.pnas.org/content/96/18/10254.full

